

今回は「報告要件」について学習しましょう。
医薬品の安全性情報報告における「報告」とは、当局に有害事象(副作用)を報告することを言います。
副作用等の報告について、医薬品医療機器法第68条の10には以下のように記載されています。
| 医薬品医療機器法第68条の10
(副作用等の報告) |
「報告」についての記載は、上記のほか、医薬品医療機器法第68条の13に、医薬品医療機器総合機構(PMDA)に情報の整理を任せることできることを記載し、実際、製薬企業などは「報告」をPMDAに行っております。
「報告」に関する詳しい説明は、医薬品医療機器法施行規則第228条の20、医薬品医療機器法施行規則第228条の23に記載されております。
報告要件
「報告要件」とは、医薬品で有害事象が発生したら、当局(医薬品医療機器総合機構:PMDA)に報告する義務が発生するかどうかを判断するための条件です。
安全性情報では、薬の種類、情報の入手元(国内か外国か)、報告の種類で「報告要件」」が変わってきます。
安全性情報報告では薬を「医薬品」と「治験薬」に区分して考えます。
では、「医薬品」とはなにか、」「治験薬」とはなにかを説明します。
「医薬品」は次の項目に」該当する薬です。
- 日本で承認・認可されたもの
- 外国で使用されている物(治験中の物も含む。)であって、当該医薬品と成分が同一のものを指し、投与経路、用法、用量、効能、効果等が異なる場合も含まれる。
- 試験研究用医薬品(製造販売後臨床試験等で使用するもの。治験薬を除く。)
- 比較臨床試験の対照薬
「治験薬」」は次の項目に」該当する薬です。
- 治験の対象とされる薬物
- 外国で使用されている物(治験中の物を含む。)であって、当該治験薬と成分が同一のものを指すものであり、投与経路、用法・容量、効能・効果等が異なる場合も含まれる。
「医薬品」と「治験薬」について、「報告要件」が報告の種類により異なり、しかもタイムフレームも異なるので、別々に数回に分けて説明することにします。
タイムフレームとは、安全性情報報告を「報告要件」に照らした結果、その報告をいつまで当局(PMDA)に報告しなければならないかを示す期日です。
それでは、「報告要件」を示して、その有害事象が報告対象になるのか、ならないのか、報告対象となるとしたらタイムフレームはどうなのかを説明した表を記載します。
医薬品の報告対象とタイムフレーム
-
- 副作用症例報告
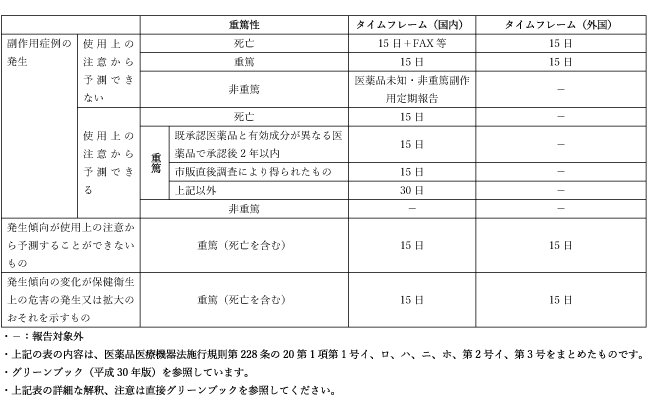
- 上記表の詳細な解釈、注意は直接グリーンブックを参照してください。
次回は「医薬品」の感染症症例報告の報告対象とタイムフレームを説明します。